Forensic Application of ForenSeqTM DNA Signature Prep Kit in Zhengjiang She Ethnic Group
QU Yi-ling,1,*, LIN Yuan,2,*, YANG Zi-hao2,3, TAO Rui-yang2, XIA Ruo-cheng2, CAO Zheng-jun4, GAO Rui-xiang4, YU Huan1,2, WANG Zi-wei1,2, YANG Qi1,2, ZHANG Xiao-chun1,2, ZHANG Su-hua,2, LI Cheng-tao,1,2
1.School of Biology and Basic Medical Sciences, Soochow University, Suzhou 215123, Jiangsu Province, China
2.Shanghai Key Laboratory of Forensic Medicine, Key Laboratory of Forensic Science, Ministry of Justice, Shanghai Forensic Service Platform, Academy of Forensic Science, Shanghai 200063, China
3.East China University of Political Science and Law, Shanghai 200042, China
4.Criminal Police Detachment of Yancheng Public Security Bureau, Yancheng 224000, Jiangsu Province, China
ObjectiveTo evaluate the ability of the ForenSeqTM DNA Signature Prep kit (ForenSeq kit) in analyzing the sequence information of STRs in Zhejiang She ethnic group and its forensic application efficacy.MethodsA total of 50 Zhejiang She ethnic group samples were sequenced with the ForenSeq kit on the MiSeq FGx platform. The data was analyzed using ForenSeqTM universal analysis software to obtain the motif structure and flank regions of the 58 STRs, then compared with PCR-CE typing results to test the consistency. At last, the allele frequency and population genetic parameters were calculated.ResultsA total of 448 sequence polymorphic alleles were detected in 50 samples of Zhejiang She ethnic group. Compared with fragment length polymorphism detected by PCR-CE, 82 alleles were increased by MPS detection based on ForenSeq kit, and 7 SNPs variation were detected in the flanking regions of 6 loci. The 22 male individuals were genotyped, and total 19 haplotypes were detected in 24 Y chromosome STRs of these 22 males. The cumulative discrimination power of the 27 autosomal STRs was 1-8.87×10-30, the cumulative probability of exclusion of duo-testing was 0.999 999 962 640 657, the cumulative probability of exclusion of trios-testing was 0.999 999 999 999 633.ConclusionBased on MPS typing technology, using the ForenSeq kit greatly improves the detection efficiency. In addition, the 58 STRs have good genetic polymorphisms in Zhejiang She ethnic group, which are suitable for individual identification and paternity identification in forensic application.
Keywords:forensic genetics
;
massively parallel sequencing
;
short tandem repeat (STR)
;
polymorphism, genetic
;
ForenSeqTM DNA Signature Prep kit
;
She ethnic group
;
Zhejiang
QU Yi-ling, LIN Yuan, YANG Zi-hao, TAO Rui-yang, XIA Ruo-cheng, CAO Zheng-jun, GAO Rui-xiang, YU Huan, WANG Zi-wei, YANG Qi, ZHANG Xiao-chun, ZHANG Su-hua, LI Cheng-tao. Forensic Application of ForenSeqTM DNA Signature Prep Kit in Zhengjiang She Ethnic Group. Journal of Forensic Medicine[J], 2021, 37(6): 817-824 DOI:10.12116/j.issn.1004-5619.2021.510404
短串联重复(short tandem repeat,STR)序列由2~6个核心重复碱基构成,是法医遗传学中应用最为广泛的DNA遗传标记[1]。大规模平行测序(massively parallel sequencing,MPS)技术也被称为下一代测序(next generation sequencing,NGS),近年来在法医学领域中的应用逐渐增多。相较于传统的聚合酶链反应-毛细管电泳(polymerase chain reaction-capillary electrophoresis,PCR-CE)分型技术,MPS技术不仅能区分等位基因片段长度大小,还可以区分等位基因的序列差异,并且能够同时对多个样本的多个遗传标记进行高覆盖度的测序分析,极大提高了法医遗传学实验室对DNA序列的识别能力和对大批量样本的处理能力[2]。
美国Illumina公司基于MiSeq FGx法医基因组学系统针对法医遗传学开发了商业化MPS检测试剂盒——ForenSeqTM DNA Signature Prep试剂盒(以下简称ForenSeq试剂盒)。该试剂盒简化了测序流程,提供了从DNA样本到数据分析的一体化解决方案,单次检测可获得59个STR和172个单核苷酸多态性(single nucleotide polymorphism,SNP)遗传标记的信息,克服了PCR-CE技术需要多个试剂盒才能检测相同数目遗传标记的难题,展现出良好的应用前景[3-5]。
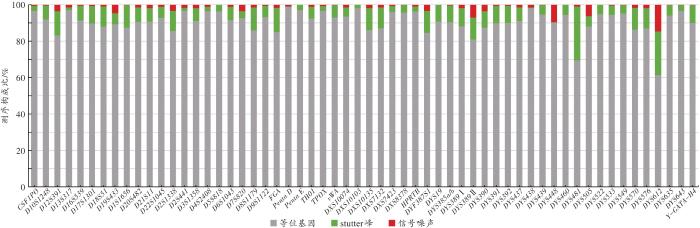
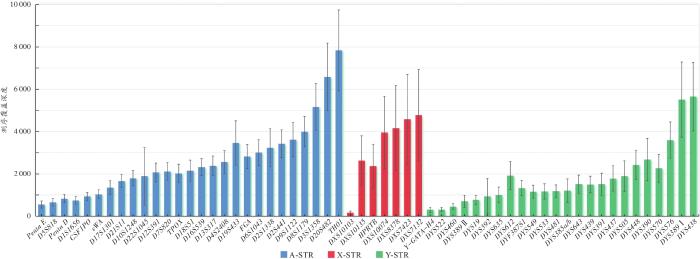
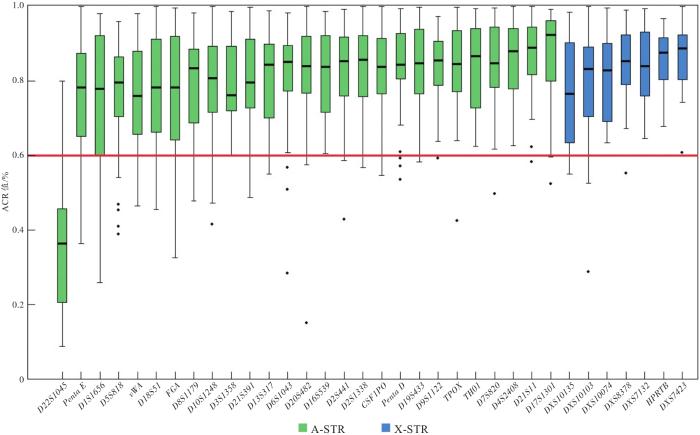
通过下机数据中的Q30值初步判断数据质量,利用ForenSeqTM通用分析软件按照默认分析阈值和解释阈值处理原始测序数据,同时使用STRait Razor v2软件[11]对测序的FASTQ文件进一步分析。从以下三方面对测序质量进行评估:(1)测序序列信息构成比,指27个常染色体STR基因座(以下简称A-STR)、7个X染色体STR基因座(以下简称X-STR)和24个Y染色体STR基因座(以下简称Y-STR)中等位基因、stutter峰和信号噪声分别占总reads的百分比;(2)测序深度;(3)等位基因覆盖率(allele coverage ratio,ACR),计算方法为用杂合子等位基因中较小的reads数除以较大的reads数。采用Arlequin v3.5软件[12]进行各基因座的Hardy-Weinberg平衡检验及基因座间的连锁不平衡检验。另外,通过与hg19(GRCh37)参考基因组进行比较,对STR基因座侧翼序列信息展开分析。最后,采用Cervus 3.0软件[13]计算各基因座的多态信息含量(polymorphic information content,PIC)、非父排除率(probability of paternity excluding,PE)、二联体非父排除率(probability of exclusion of duo-testing,PEduo)、三联体非父排除率(probability of exclusion of trios-testing,PEtrio)、平均非父排除率(mean probability of exclusion)等,运用群体遗传分析软件[13]计算各基因座的个体识别率(discrimination power,DP),依据《常染色体STR基因座的法医学参数计算规范》(SF/Z JD0105010—2018)计算累积个体识别率(cumulative discrimination power,CDP)、累积非父排除率(cumulative probability of exclusion,CPE)和累积平均非父排除率(cumulative mean probability of exclusion),依据《法医物证鉴定Y-STR检验规范》(SF/Z JD0105007—2018)计算基因多样性(gene diversity,GD)、单倍型多样性(haplotype diversity,HD)、单倍型的分辨能力(discrimination capacity,DC)和单倍型匹配概率(haplotype match probability,HMP)。
基于PCR-CE技术对50例畲族个体血样的58个STR基因座进行检测,分型结果与MPS检测结果一致。此外,在MPS数据分析过程中发现,ForenSeq试剂盒所提供的部分STR基因座核心序列信息与国际法医遗传学会(International Society for Forensic Genetics,ISFG)对MPS-STR的命名建议[14]存在出入,其中D1S1656、D2S1338、D5S818、CSF1P0、D6S1043、D7S820、Penta E、D19S433、FGA、HPRTB、DXS8378、DXS7423、DYS19、DYS389Ⅰ、DYS389Ⅱ、DYS390、DYS392与DYS460基因座的核心序列结构信息与ISFG建议不一致。
Massively parallel sequencing of forensic STRs and SNPs using the Illumina® ForenSeqTM DNA Signature Prep Kit on the MiSeq FGxTM Forensic Genomics System
Population genetic analysis of 36 Y-chromosomal STRs yields comprehensive insights into the forensic features and phylogenetic relationship of Chinese Tai-Kadai-speaking Bouyei
Massively parallel sequencing of forensic STRs: Considerations of the DNA commission of the International Society for Forensic Genetics (ISFG) on minimal nomenclature requirements
Massively parallel sequencing of forensic STRs and SNPs using the Illumina? ForenSeqTM DNA Signature Prep Kit on the MiSeq FGxTM Forensic Genomics System
2
2017
... 美国Illumina公司基于MiSeq FGx法医基因组学系统针对法医遗传学开发了商业化MPS检测试剂盒——ForenSeqTM DNA Signature Prep试剂盒(以下简称ForenSeq试剂盒).该试剂盒简化了测序流程,提供了从DNA样本到数据分析的一体化解决方案,单次检测可获得59个STR和172个单核苷酸多态性(single nucleotide polymorphism,SNP)遗传标记的信息,克服了PCR-CE技术需要多个试剂盒才能检测相同数目遗传标记的难题,展现出良好的应用前景[3-5]. ...
Population genetic analysis of 36 Y-chromosomal STRs yields comprehensive insights into the forensic features and phylogenetic relationship of Chinese Tai-Kadai-speaking Bouyei
STRait Razor: A length-based forensic STR allele-calling tool for use with second generation sequencing data
1
2013
... 通过下机数据中的Q30值初步判断数据质量,利用ForenSeqTM通用分析软件按照默认分析阈值和解释阈值处理原始测序数据,同时使用STRait Razor v2软件[11]对测序的FASTQ文件进一步分析.从以下三方面对测序质量进行评估:(1)测序序列信息构成比,指27个常染色体STR基因座(以下简称A-STR)、7个X染色体STR基因座(以下简称X-STR)和24个Y染色体STR基因座(以下简称Y-STR)中等位基因、stutter峰和信号噪声分别占总reads的百分比;(2)测序深度;(3)等位基因覆盖率(allele coverage ratio,ACR),计算方法为用杂合子等位基因中较小的reads数除以较大的reads数.采用Arlequin v3.5软件[12]进行各基因座的Hardy-Weinberg平衡检验及基因座间的连锁不平衡检验.另外,通过与hg19(GRCh37)参考基因组进行比较,对STR基因座侧翼序列信息展开分析.最后,采用Cervus 3.0软件[13]计算各基因座的多态信息含量(polymorphic information content,PIC)、非父排除率(probability of paternity excluding,PE)、二联体非父排除率(probability of exclusion of duo-testing,PEduo)、三联体非父排除率(probability of exclusion of trios-testing,PEtrio)、平均非父排除率(mean probability of exclusion)等,运用群体遗传分析软件[13]计算各基因座的个体识别率(discrimination power,DP),依据《常染色体STR基因座的法医学参数计算规范》(SF/Z JD0105010—2018)计算累积个体识别率(cumulative discrimination power,CDP)、累积非父排除率(cumulative probability of exclusion,CPE)和累积平均非父排除率(cumulative mean probability of exclusion),依据《法医物证鉴定Y-STR检验规范》(SF/Z JD0105007—2018)计算基因多样性(gene diversity,GD)、单倍型多样性(haplotype diversity,HD)、单倍型的分辨能力(discrimination capacity,DC)和单倍型匹配概率(haplotype match probability,HMP). ...
Arlequin suite ver 3.5: A new series of programs to perform population genetics analyses under Linux and Windows
1
2010
... 通过下机数据中的Q30值初步判断数据质量,利用ForenSeqTM通用分析软件按照默认分析阈值和解释阈值处理原始测序数据,同时使用STRait Razor v2软件[11]对测序的FASTQ文件进一步分析.从以下三方面对测序质量进行评估:(1)测序序列信息构成比,指27个常染色体STR基因座(以下简称A-STR)、7个X染色体STR基因座(以下简称X-STR)和24个Y染色体STR基因座(以下简称Y-STR)中等位基因、stutter峰和信号噪声分别占总reads的百分比;(2)测序深度;(3)等位基因覆盖率(allele coverage ratio,ACR),计算方法为用杂合子等位基因中较小的reads数除以较大的reads数.采用Arlequin v3.5软件[12]进行各基因座的Hardy-Weinberg平衡检验及基因座间的连锁不平衡检验.另外,通过与hg19(GRCh37)参考基因组进行比较,对STR基因座侧翼序列信息展开分析.最后,采用Cervus 3.0软件[13]计算各基因座的多态信息含量(polymorphic information content,PIC)、非父排除率(probability of paternity excluding,PE)、二联体非父排除率(probability of exclusion of duo-testing,PEduo)、三联体非父排除率(probability of exclusion of trios-testing,PEtrio)、平均非父排除率(mean probability of exclusion)等,运用群体遗传分析软件[13]计算各基因座的个体识别率(discrimination power,DP),依据《常染色体STR基因座的法医学参数计算规范》(SF/Z JD0105010—2018)计算累积个体识别率(cumulative discrimination power,CDP)、累积非父排除率(cumulative probability of exclusion,CPE)和累积平均非父排除率(cumulative mean probability of exclusion),依据《法医物证鉴定Y-STR检验规范》(SF/Z JD0105007—2018)计算基因多样性(gene diversity,GD)、单倍型多样性(haplotype diversity,HD)、单倍型的分辨能力(discrimination capacity,DC)和单倍型匹配概率(haplotype match probability,HMP). ...
几款遗传学分析软件在法医生物统计中的应用
2
2014
... 通过下机数据中的Q30值初步判断数据质量,利用ForenSeqTM通用分析软件按照默认分析阈值和解释阈值处理原始测序数据,同时使用STRait Razor v2软件[11]对测序的FASTQ文件进一步分析.从以下三方面对测序质量进行评估:(1)测序序列信息构成比,指27个常染色体STR基因座(以下简称A-STR)、7个X染色体STR基因座(以下简称X-STR)和24个Y染色体STR基因座(以下简称Y-STR)中等位基因、stutter峰和信号噪声分别占总reads的百分比;(2)测序深度;(3)等位基因覆盖率(allele coverage ratio,ACR),计算方法为用杂合子等位基因中较小的reads数除以较大的reads数.采用Arlequin v3.5软件[12]进行各基因座的Hardy-Weinberg平衡检验及基因座间的连锁不平衡检验.另外,通过与hg19(GRCh37)参考基因组进行比较,对STR基因座侧翼序列信息展开分析.最后,采用Cervus 3.0软件[13]计算各基因座的多态信息含量(polymorphic information content,PIC)、非父排除率(probability of paternity excluding,PE)、二联体非父排除率(probability of exclusion of duo-testing,PEduo)、三联体非父排除率(probability of exclusion of trios-testing,PEtrio)、平均非父排除率(mean probability of exclusion)等,运用群体遗传分析软件[13]计算各基因座的个体识别率(discrimination power,DP),依据《常染色体STR基因座的法医学参数计算规范》(SF/Z JD0105010—2018)计算累积个体识别率(cumulative discrimination power,CDP)、累积非父排除率(cumulative probability of exclusion,CPE)和累积平均非父排除率(cumulative mean probability of exclusion),依据《法医物证鉴定Y-STR检验规范》(SF/Z JD0105007—2018)计算基因多样性(gene diversity,GD)、单倍型多样性(haplotype diversity,HD)、单倍型的分辨能力(discrimination capacity,DC)和单倍型匹配概率(haplotype match probability,HMP). ...
... [13]计算各基因座的个体识别率(discrimination power,DP),依据《常染色体STR基因座的法医学参数计算规范》(SF/Z JD0105010—2018)计算累积个体识别率(cumulative discrimination power,CDP)、累积非父排除率(cumulative probability of exclusion,CPE)和累积平均非父排除率(cumulative mean probability of exclusion),依据《法医物证鉴定Y-STR检验规范》(SF/Z JD0105007—2018)计算基因多样性(gene diversity,GD)、单倍型多样性(haplotype diversity,HD)、单倍型的分辨能力(discrimination capacity,DC)和单倍型匹配概率(haplotype match probability,HMP). ...
几款遗传学分析软件在法医生物统计中的应用
2
2014
... 通过下机数据中的Q30值初步判断数据质量,利用ForenSeqTM通用分析软件按照默认分析阈值和解释阈值处理原始测序数据,同时使用STRait Razor v2软件[11]对测序的FASTQ文件进一步分析.从以下三方面对测序质量进行评估:(1)测序序列信息构成比,指27个常染色体STR基因座(以下简称A-STR)、7个X染色体STR基因座(以下简称X-STR)和24个Y染色体STR基因座(以下简称Y-STR)中等位基因、stutter峰和信号噪声分别占总reads的百分比;(2)测序深度;(3)等位基因覆盖率(allele coverage ratio,ACR),计算方法为用杂合子等位基因中较小的reads数除以较大的reads数.采用Arlequin v3.5软件[12]进行各基因座的Hardy-Weinberg平衡检验及基因座间的连锁不平衡检验.另外,通过与hg19(GRCh37)参考基因组进行比较,对STR基因座侧翼序列信息展开分析.最后,采用Cervus 3.0软件[13]计算各基因座的多态信息含量(polymorphic information content,PIC)、非父排除率(probability of paternity excluding,PE)、二联体非父排除率(probability of exclusion of duo-testing,PEduo)、三联体非父排除率(probability of exclusion of trios-testing,PEtrio)、平均非父排除率(mean probability of exclusion)等,运用群体遗传分析软件[13]计算各基因座的个体识别率(discrimination power,DP),依据《常染色体STR基因座的法医学参数计算规范》(SF/Z JD0105010—2018)计算累积个体识别率(cumulative discrimination power,CDP)、累积非父排除率(cumulative probability of exclusion,CPE)和累积平均非父排除率(cumulative mean probability of exclusion),依据《法医物证鉴定Y-STR检验规范》(SF/Z JD0105007—2018)计算基因多样性(gene diversity,GD)、单倍型多样性(haplotype diversity,HD)、单倍型的分辨能力(discrimination capacity,DC)和单倍型匹配概率(haplotype match probability,HMP). ...
... [13]计算各基因座的个体识别率(discrimination power,DP),依据《常染色体STR基因座的法医学参数计算规范》(SF/Z JD0105010—2018)计算累积个体识别率(cumulative discrimination power,CDP)、累积非父排除率(cumulative probability of exclusion,CPE)和累积平均非父排除率(cumulative mean probability of exclusion),依据《法医物证鉴定Y-STR检验规范》(SF/Z JD0105007—2018)计算基因多样性(gene diversity,GD)、单倍型多样性(haplotype diversity,HD)、单倍型的分辨能力(discrimination capacity,DC)和单倍型匹配概率(haplotype match probability,HMP). ...
Massively parallel sequencing of forensic STRs: Considerations of the DNA commission of the International Society for Forensic Genetics (ISFG) on minimal nomenclature requirements
2
2016
... 基于PCR-CE技术对50例畲族个体血样的58个STR基因座进行检测,分型结果与MPS检测结果一致.此外,在MPS数据分析过程中发现,ForenSeq试剂盒所提供的部分STR基因座核心序列信息与国际法医遗传学会(International Society for Forensic Genetics,ISFG)对MPS-STR的命名建议[14]存在出入,其中D1S1656、D2S1338、D5S818、CSF1P0、D6S1043、D7S820、Penta E、D19S433、FGA、HPRTB、DXS8378、DXS7423、DYS19、DYS389Ⅰ、DYS389Ⅱ、DYS390、DYS392与DYS460基因座的核心序列结构信息与ISFG建议不一致. ...



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}