人体微生物组研究进展
1
2020
... 细菌是人体的重要组成部分,在人体各部位形成复杂多样的微生态系统[1-2].由于性别、年龄、生活环境、饮食习惯、健康状况等的不同,人体菌群具有个体特异性,同一个体不同部位的菌群也不相同[3].人体菌群很容易转移到现场物品上,或随分泌物、排泄物遗留在现场,甚至比人体细胞的数量还多,且能在一定时间内保持菌群原有的特性[4].人死后,尸体各处的菌群随腐败进展而发生有规律的变化[5-6].现场检材或尸体的菌群特征可提供斑痕类型、来源个体及死亡时间等信息[7-8],菌群分析作为解决法医学问题的新途径日益受到关注. ...
人体微生物组研究进展
1
2020
... 细菌是人体的重要组成部分,在人体各部位形成复杂多样的微生态系统[1-2].由于性别、年龄、生活环境、饮食习惯、健康状况等的不同,人体菌群具有个体特异性,同一个体不同部位的菌群也不相同[3].人体菌群很容易转移到现场物品上,或随分泌物、排泄物遗留在现场,甚至比人体细胞的数量还多,且能在一定时间内保持菌群原有的特性[4].人死后,尸体各处的菌群随腐败进展而发生有规律的变化[5-6].现场检材或尸体的菌群特征可提供斑痕类型、来源个体及死亡时间等信息[7-8],菌群分析作为解决法医学问题的新途径日益受到关注. ...
Individuality of the composition of the human microbiota
1
2021
... 细菌是人体的重要组成部分,在人体各部位形成复杂多样的微生态系统[1-2].由于性别、年龄、生活环境、饮食习惯、健康状况等的不同,人体菌群具有个体特异性,同一个体不同部位的菌群也不相同[3].人体菌群很容易转移到现场物品上,或随分泌物、排泄物遗留在现场,甚至比人体细胞的数量还多,且能在一定时间内保持菌群原有的特性[4].人死后,尸体各处的菌群随腐败进展而发生有规律的变化[5-6].现场检材或尸体的菌群特征可提供斑痕类型、来源个体及死亡时间等信息[7-8],菌群分析作为解决法医学问题的新途径日益受到关注. ...
皮肤微生物群落的研究进展及法医学应用
1
2019
... 细菌是人体的重要组成部分,在人体各部位形成复杂多样的微生态系统[1-2].由于性别、年龄、生活环境、饮食习惯、健康状况等的不同,人体菌群具有个体特异性,同一个体不同部位的菌群也不相同[3].人体菌群很容易转移到现场物品上,或随分泌物、排泄物遗留在现场,甚至比人体细胞的数量还多,且能在一定时间内保持菌群原有的特性[4].人死后,尸体各处的菌群随腐败进展而发生有规律的变化[5-6].现场检材或尸体的菌群特征可提供斑痕类型、来源个体及死亡时间等信息[7-8],菌群分析作为解决法医学问题的新途径日益受到关注. ...
皮肤微生物群落的研究进展及法医学应用
1
2019
... 细菌是人体的重要组成部分,在人体各部位形成复杂多样的微生态系统[1-2].由于性别、年龄、生活环境、饮食习惯、健康状况等的不同,人体菌群具有个体特异性,同一个体不同部位的菌群也不相同[3].人体菌群很容易转移到现场物品上,或随分泌物、排泄物遗留在现场,甚至比人体细胞的数量还多,且能在一定时间内保持菌群原有的特性[4].人死后,尸体各处的菌群随腐败进展而发生有规律的变化[5-6].现场检材或尸体的菌群特征可提供斑痕类型、来源个体及死亡时间等信息[7-8],菌群分析作为解决法医学问题的新途径日益受到关注. ...
Challenges in human skin microbial profiling for forensic science: A review
2
2020
... 细菌是人体的重要组成部分,在人体各部位形成复杂多样的微生态系统[1-2].由于性别、年龄、生活环境、饮食习惯、健康状况等的不同,人体菌群具有个体特异性,同一个体不同部位的菌群也不相同[3].人体菌群很容易转移到现场物品上,或随分泌物、排泄物遗留在现场,甚至比人体细胞的数量还多,且能在一定时间内保持菌群原有的特性[4].人死后,尸体各处的菌群随腐败进展而发生有规律的变化[5-6].现场检材或尸体的菌群特征可提供斑痕类型、来源个体及死亡时间等信息[7-8],菌群分析作为解决法医学问题的新途径日益受到关注. ...
... 菌群可用形态、生物化学和遗传学等方法进行分析,但受检材条件等限制,目前法医学菌群分析主要指菌群的DNA分析,常用的标记基因是以序列变异为主的16S rDNA,分析过程一般包括检材处理、DNA提取、扩增、PCR产物检测和数据分析等多个环节[4-10].在DNA提取环节,又有裂解、纯化等步骤,耗时长,检材用量大.PCR产物则须用杂交、测序或梯度凝胶电泳等特殊的技术进行分离、检测,操作繁琐,成本高,污染风险大.此外,菌群PCR是一种以群体特征为研究对象的多模板PCR[11],不同于法医学常用的多重PCR,不能按照传统的PCR来优化和评价.现有的菌群分析技术均来自其他领域,不符合法医学工作特点,也未经改造和验证,难以满足法医学检验的要求. ...
Biolog-Eco法检测尸体微生物群落的代谢功能变化
1
2016
... 细菌是人体的重要组成部分,在人体各部位形成复杂多样的微生态系统[1-2].由于性别、年龄、生活环境、饮食习惯、健康状况等的不同,人体菌群具有个体特异性,同一个体不同部位的菌群也不相同[3].人体菌群很容易转移到现场物品上,或随分泌物、排泄物遗留在现场,甚至比人体细胞的数量还多,且能在一定时间内保持菌群原有的特性[4].人死后,尸体各处的菌群随腐败进展而发生有规律的变化[5-6].现场检材或尸体的菌群特征可提供斑痕类型、来源个体及死亡时间等信息[7-8],菌群分析作为解决法医学问题的新途径日益受到关注. ...
Biolog-Eco法检测尸体微生物群落的代谢功能变化
1
2016
... 细菌是人体的重要组成部分,在人体各部位形成复杂多样的微生态系统[1-2].由于性别、年龄、生活环境、饮食习惯、健康状况等的不同,人体菌群具有个体特异性,同一个体不同部位的菌群也不相同[3].人体菌群很容易转移到现场物品上,或随分泌物、排泄物遗留在现场,甚至比人体细胞的数量还多,且能在一定时间内保持菌群原有的特性[4].人死后,尸体各处的菌群随腐败进展而发生有规律的变化[5-6].现场检材或尸体的菌群特征可提供斑痕类型、来源个体及死亡时间等信息[7-8],菌群分析作为解决法医学问题的新途径日益受到关注. ...
泥土微生物检验及其在法庭科学中的应用
1
2019
... 细菌是人体的重要组成部分,在人体各部位形成复杂多样的微生态系统[1-2].由于性别、年龄、生活环境、饮食习惯、健康状况等的不同,人体菌群具有个体特异性,同一个体不同部位的菌群也不相同[3].人体菌群很容易转移到现场物品上,或随分泌物、排泄物遗留在现场,甚至比人体细胞的数量还多,且能在一定时间内保持菌群原有的特性[4].人死后,尸体各处的菌群随腐败进展而发生有规律的变化[5-6].现场检材或尸体的菌群特征可提供斑痕类型、来源个体及死亡时间等信息[7-8],菌群分析作为解决法医学问题的新途径日益受到关注. ...
泥土微生物检验及其在法庭科学中的应用
1
2019
... 细菌是人体的重要组成部分,在人体各部位形成复杂多样的微生态系统[1-2].由于性别、年龄、生活环境、饮食习惯、健康状况等的不同,人体菌群具有个体特异性,同一个体不同部位的菌群也不相同[3].人体菌群很容易转移到现场物品上,或随分泌物、排泄物遗留在现场,甚至比人体细胞的数量还多,且能在一定时间内保持菌群原有的特性[4].人死后,尸体各处的菌群随腐败进展而发生有规律的变化[5-6].现场检材或尸体的菌群特征可提供斑痕类型、来源个体及死亡时间等信息[7-8],菌群分析作为解决法医学问题的新途径日益受到关注. ...
宏基因组学在法医学鉴定中的应用
1
2017
... 细菌是人体的重要组成部分,在人体各部位形成复杂多样的微生态系统[1-2].由于性别、年龄、生活环境、饮食习惯、健康状况等的不同,人体菌群具有个体特异性,同一个体不同部位的菌群也不相同[3].人体菌群很容易转移到现场物品上,或随分泌物、排泄物遗留在现场,甚至比人体细胞的数量还多,且能在一定时间内保持菌群原有的特性[4].人死后,尸体各处的菌群随腐败进展而发生有规律的变化[5-6].现场检材或尸体的菌群特征可提供斑痕类型、来源个体及死亡时间等信息[7-8],菌群分析作为解决法医学问题的新途径日益受到关注. ...
3969/j.issn.1004-5619.2017.04.014. SA R N, CAI L Y, WU H J,
1
2017
... 菌群可用形态、生物化学和遗传学等方法进行分析,但受检材条件等限制,目前法医学菌群分析主要指菌群的DNA分析,常用的标记基因是以序列变异为主的16S rDNA,分析过程一般包括检材处理、DNA提取、扩增、PCR产物检测和数据分析等多个环节[4-10].在DNA提取环节,又有裂解、纯化等步骤,耗时长,检材用量大.PCR产物则须用杂交、测序或梯度凝胶电泳等特殊的技术进行分离、检测,操作繁琐,成本高,污染风险大.此外,菌群PCR是一种以群体特征为研究对象的多模板PCR[11],不同于法医学常用的多重PCR,不能按照传统的PCR来优化和评价.现有的菌群分析技术均来自其他领域,不符合法医学工作特点,也未经改造和验证,难以满足法医学检验的要求. ...
16S rRNA基因测序在法医学中的研究进展
1
2018
... 细菌是人体的重要组成部分,在人体各部位形成复杂多样的微生态系统[1-2].由于性别、年龄、生活环境、饮食习惯、健康状况等的不同,人体菌群具有个体特异性,同一个体不同部位的菌群也不相同[3].人体菌群很容易转移到现场物品上,或随分泌物、排泄物遗留在现场,甚至比人体细胞的数量还多,且能在一定时间内保持菌群原有的特性[4].人死后,尸体各处的菌群随腐败进展而发生有规律的变化[5-6].现场检材或尸体的菌群特征可提供斑痕类型、来源个体及死亡时间等信息[7-8],菌群分析作为解决法医学问题的新途径日益受到关注. ...
16S rRNA基因测序在法医学中的研究进展
1
2018
... 细菌是人体的重要组成部分,在人体各部位形成复杂多样的微生态系统[1-2].由于性别、年龄、生活环境、饮食习惯、健康状况等的不同,人体菌群具有个体特异性,同一个体不同部位的菌群也不相同[3].人体菌群很容易转移到现场物品上,或随分泌物、排泄物遗留在现场,甚至比人体细胞的数量还多,且能在一定时间内保持菌群原有的特性[4].人死后,尸体各处的菌群随腐败进展而发生有规律的变化[5-6].现场检材或尸体的菌群特征可提供斑痕类型、来源个体及死亡时间等信息[7-8],菌群分析作为解决法医学问题的新途径日益受到关注. ...
示指与接触物菌群ERIC-PCR指纹图谱比对
1
2018
... 菌群检测在PCR后须用变性梯度凝胶电泳(denaturing gradient gel electrophoresis,DGGE)等较为复杂的方法进行分析,耗时费力[9-11].双链DNA的熔解曲线因长度、序列和GC含量而异,HRM利用解链温度差异鉴别DNA,为菌群PCR产物的分析提供了一种新途径.可在扩增前将EvaGreen荧光染料加入PCR体系,连续进行扩增和PCR产物分析.菌群的16S rDNA片段扩增后,体系内既有来自各种细菌同一基因但序列各异的同源双链DNA,也有这些序列相互杂交形成的、不同程度错配的异源双链DNA.这些同源、异源双链DNA的解链温度各不相同,拷贝数也因样本的菌群结构而异,整个体系在热变性时会生成复杂的熔解曲线,具体形态取决于样本的菌群结构,即细菌的种类和各种细菌的丰度.HRM软件把每个样本的曲线作为一个整体进行分析、比较,计算样本曲线之间的欧氏距离,转换成GCP,以客观、量化的形式反映两个样本HRM曲线的相似度. ...
示指与接触物菌群ERIC-PCR指纹图谱比对
1
2018
... 菌群检测在PCR后须用变性梯度凝胶电泳(denaturing gradient gel electrophoresis,DGGE)等较为复杂的方法进行分析,耗时费力[9-11].双链DNA的熔解曲线因长度、序列和GC含量而异,HRM利用解链温度差异鉴别DNA,为菌群PCR产物的分析提供了一种新途径.可在扩增前将EvaGreen荧光染料加入PCR体系,连续进行扩增和PCR产物分析.菌群的16S rDNA片段扩增后,体系内既有来自各种细菌同一基因但序列各异的同源双链DNA,也有这些序列相互杂交形成的、不同程度错配的异源双链DNA.这些同源、异源双链DNA的解链温度各不相同,拷贝数也因样本的菌群结构而异,整个体系在热变性时会生成复杂的熔解曲线,具体形态取决于样本的菌群结构,即细菌的种类和各种细菌的丰度.HRM软件把每个样本的曲线作为一个整体进行分析、比较,计算样本曲线之间的欧氏距离,转换成GCP,以客观、量化的形式反映两个样本HRM曲线的相似度. ...
土壤微生物16S rDNA的T-RFLP法医学应用分析
2
2016
... 菌群可用形态、生物化学和遗传学等方法进行分析,但受检材条件等限制,目前法医学菌群分析主要指菌群的DNA分析,常用的标记基因是以序列变异为主的16S rDNA,分析过程一般包括检材处理、DNA提取、扩增、PCR产物检测和数据分析等多个环节[4-10].在DNA提取环节,又有裂解、纯化等步骤,耗时长,检材用量大.PCR产物则须用杂交、测序或梯度凝胶电泳等特殊的技术进行分离、检测,操作繁琐,成本高,污染风险大.此外,菌群PCR是一种以群体特征为研究对象的多模板PCR[11],不同于法医学常用的多重PCR,不能按照传统的PCR来优化和评价.现有的菌群分析技术均来自其他领域,不符合法医学工作特点,也未经改造和验证,难以满足法医学检验的要求. ...
... 菌群检测在PCR后须用变性梯度凝胶电泳(denaturing gradient gel electrophoresis,DGGE)等较为复杂的方法进行分析,耗时费力[9-11].双链DNA的熔解曲线因长度、序列和GC含量而异,HRM利用解链温度差异鉴别DNA,为菌群PCR产物的分析提供了一种新途径.可在扩增前将EvaGreen荧光染料加入PCR体系,连续进行扩增和PCR产物分析.菌群的16S rDNA片段扩增后,体系内既有来自各种细菌同一基因但序列各异的同源双链DNA,也有这些序列相互杂交形成的、不同程度错配的异源双链DNA.这些同源、异源双链DNA的解链温度各不相同,拷贝数也因样本的菌群结构而异,整个体系在热变性时会生成复杂的熔解曲线,具体形态取决于样本的菌群结构,即细菌的种类和各种细菌的丰度.HRM软件把每个样本的曲线作为一个整体进行分析、比较,计算样本曲线之间的欧氏距离,转换成GCP,以客观、量化的形式反映两个样本HRM曲线的相似度. ...
土壤微生物16S rDNA的T-RFLP法医学应用分析
2
2016
... 菌群可用形态、生物化学和遗传学等方法进行分析,但受检材条件等限制,目前法医学菌群分析主要指菌群的DNA分析,常用的标记基因是以序列变异为主的16S rDNA,分析过程一般包括检材处理、DNA提取、扩增、PCR产物检测和数据分析等多个环节[4-10].在DNA提取环节,又有裂解、纯化等步骤,耗时长,检材用量大.PCR产物则须用杂交、测序或梯度凝胶电泳等特殊的技术进行分离、检测,操作繁琐,成本高,污染风险大.此外,菌群PCR是一种以群体特征为研究对象的多模板PCR[11],不同于法医学常用的多重PCR,不能按照传统的PCR来优化和评价.现有的菌群分析技术均来自其他领域,不符合法医学工作特点,也未经改造和验证,难以满足法医学检验的要求. ...
... 菌群检测在PCR后须用变性梯度凝胶电泳(denaturing gradient gel electrophoresis,DGGE)等较为复杂的方法进行分析,耗时费力[9-11].双链DNA的熔解曲线因长度、序列和GC含量而异,HRM利用解链温度差异鉴别DNA,为菌群PCR产物的分析提供了一种新途径.可在扩增前将EvaGreen荧光染料加入PCR体系,连续进行扩增和PCR产物分析.菌群的16S rDNA片段扩增后,体系内既有来自各种细菌同一基因但序列各异的同源双链DNA,也有这些序列相互杂交形成的、不同程度错配的异源双链DNA.这些同源、异源双链DNA的解链温度各不相同,拷贝数也因样本的菌群结构而异,整个体系在热变性时会生成复杂的熔解曲线,具体形态取决于样本的菌群结构,即细菌的种类和各种细菌的丰度.HRM软件把每个样本的曲线作为一个整体进行分析、比较,计算样本曲线之间的欧氏距离,转换成GCP,以客观、量化的形式反映两个样本HRM曲线的相似度. ...
Multi-template polymerase chain reaction
1
2014
... 熔解曲线分析法利用解链温度差异来鉴别DNA,在荧光定量PCR仪上扩增的DNA可直接进行熔解曲线分析[12].使用EvaGreen等饱和荧光染料和高精度的PCR仪,可以提高熔解曲线的分辨率,即高分辨率熔解(high resolution melting,HRM).HRM过程简单、快速,灵敏度和信息含量高,已应用于变异或突变筛查、病原体快速检测、法医DNA分型等[13-16].关于HRM在菌群分析中的应用,EVERMAN等[17]已有初步探索,WANG等[18]也已将其用于人类唾液菌群的比较,但都要先提取菌群的宏基因组DNA.直接PCR(direct PCR,dPCR)扩增已是成熟的法医DNA分型技术,在菌落筛选、病原体诊断中也有应用[19],可以简化操作、缩短时间,是菌群DNA分析快速化的另一潜在途径.16S rDNA是菌群分析中最常用的标记基因,其V4高变区在进化关系上更接近16S rDNA全长,通用性和分辨率均较高,且在法医学领域已有报道[18,20]. ...
单管一步甲基化可变位点分析技术
1
2013
... 熔解曲线分析法利用解链温度差异来鉴别DNA,在荧光定量PCR仪上扩增的DNA可直接进行熔解曲线分析[12].使用EvaGreen等饱和荧光染料和高精度的PCR仪,可以提高熔解曲线的分辨率,即高分辨率熔解(high resolution melting,HRM).HRM过程简单、快速,灵敏度和信息含量高,已应用于变异或突变筛查、病原体快速检测、法医DNA分型等[13-16].关于HRM在菌群分析中的应用,EVERMAN等[17]已有初步探索,WANG等[18]也已将其用于人类唾液菌群的比较,但都要先提取菌群的宏基因组DNA.直接PCR(direct PCR,dPCR)扩增已是成熟的法医DNA分型技术,在菌落筛选、病原体诊断中也有应用[19],可以简化操作、缩短时间,是菌群DNA分析快速化的另一潜在途径.16S rDNA是菌群分析中最常用的标记基因,其V4高变区在进化关系上更接近16S rDNA全长,通用性和分辨率均较高,且在法医学领域已有报道[18,20]. ...
单管一步甲基化可变位点分析技术
1
2013
... 熔解曲线分析法利用解链温度差异来鉴别DNA,在荧光定量PCR仪上扩增的DNA可直接进行熔解曲线分析[12].使用EvaGreen等饱和荧光染料和高精度的PCR仪,可以提高熔解曲线的分辨率,即高分辨率熔解(high resolution melting,HRM).HRM过程简单、快速,灵敏度和信息含量高,已应用于变异或突变筛查、病原体快速检测、法医DNA分型等[13-16].关于HRM在菌群分析中的应用,EVERMAN等[17]已有初步探索,WANG等[18]也已将其用于人类唾液菌群的比较,但都要先提取菌群的宏基因组DNA.直接PCR(direct PCR,dPCR)扩增已是成熟的法医DNA分型技术,在菌落筛选、病原体诊断中也有应用[19],可以简化操作、缩短时间,是菌群DNA分析快速化的另一潜在途径.16S rDNA是菌群分析中最常用的标记基因,其V4高变区在进化关系上更接近16S rDNA全长,通用性和分辨率均较高,且在法医学领域已有报道[18,20]. ...
High-resolution melting for detecting KRAS mutations in colorectal cancer
0
2019
Investigation of screening method for DNMT3A mutations by high-resolution melting analysis in acute myeloid leukemia
0
2019
Genotyping DNA variants with high-resolution melting analysis
1
2017
... 熔解曲线分析法利用解链温度差异来鉴别DNA,在荧光定量PCR仪上扩增的DNA可直接进行熔解曲线分析[12].使用EvaGreen等饱和荧光染料和高精度的PCR仪,可以提高熔解曲线的分辨率,即高分辨率熔解(high resolution melting,HRM).HRM过程简单、快速,灵敏度和信息含量高,已应用于变异或突变筛查、病原体快速检测、法医DNA分型等[13-16].关于HRM在菌群分析中的应用,EVERMAN等[17]已有初步探索,WANG等[18]也已将其用于人类唾液菌群的比较,但都要先提取菌群的宏基因组DNA.直接PCR(direct PCR,dPCR)扩增已是成熟的法医DNA分型技术,在菌落筛选、病原体诊断中也有应用[19],可以简化操作、缩短时间,是菌群DNA分析快速化的另一潜在途径.16S rDNA是菌群分析中最常用的标记基因,其V4高变区在进化关系上更接近16S rDNA全长,通用性和分辨率均较高,且在法医学领域已有报道[18,20]. ...
High resolution melting analysis for gene scanning
2
2010
... 熔解曲线分析法利用解链温度差异来鉴别DNA,在荧光定量PCR仪上扩增的DNA可直接进行熔解曲线分析[12].使用EvaGreen等饱和荧光染料和高精度的PCR仪,可以提高熔解曲线的分辨率,即高分辨率熔解(high resolution melting,HRM).HRM过程简单、快速,灵敏度和信息含量高,已应用于变异或突变筛查、病原体快速检测、法医DNA分型等[13-16].关于HRM在菌群分析中的应用,EVERMAN等[17]已有初步探索,WANG等[18]也已将其用于人类唾液菌群的比较,但都要先提取菌群的宏基因组DNA.直接PCR(direct PCR,dPCR)扩增已是成熟的法医DNA分型技术,在菌落筛选、病原体诊断中也有应用[19],可以简化操作、缩短时间,是菌群DNA分析快速化的另一潜在途径.16S rDNA是菌群分析中最常用的标记基因,其V4高变区在进化关系上更接近16S rDNA全长,通用性和分辨率均较高,且在法医学领域已有报道[18,20]. ...
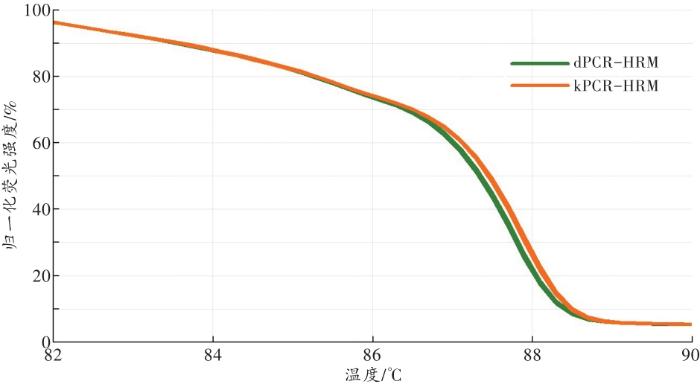
... 近年已有使用dPCR快速分析细菌DNA的报道[31],但均以特定细菌为目标.与单一序列的扩增不同,菌群PCR不仅需要样本释放足够DNA模板,还应尽量保持各种细菌DNA所占的比例.为了验证dPCR用于菌群分析的可行性,本研究选取5份唾液样本,每份同时使用dPCR-HRM和kPCR-HRM进行菌群分析.两种方法除模板不同外,其他条件保持一致.结果表明,只要两种方法所用模板的样本来源相同,得到的HRM图谱高度相似,GCP超过80%[GCP>95.85%(96.95%±1.56%)].这与多数文献[19,22,28]报道的一致,即提取方法对菌群DNA分析的影响较小.至少对于唾液样本的菌群分型来说,不论直接扩增还是提取后扩增,两者的有效模板构成基本相同,不影响其HRM型的判读.HRM技术用于菌群分析的可行性已被EVERMAN等[17,32]的研究证实,免于提取的dPCR-HRM在此基础上进一步简化,可在90 min内同步完成数十甚至上百个样本的分析,同时也更便于操作的自动化. ...
Rapid differentiation of bacterial communities using high resolution melting analysis
3
2017
... 熔解曲线分析法利用解链温度差异来鉴别DNA,在荧光定量PCR仪上扩增的DNA可直接进行熔解曲线分析[12].使用EvaGreen等饱和荧光染料和高精度的PCR仪,可以提高熔解曲线的分辨率,即高分辨率熔解(high resolution melting,HRM).HRM过程简单、快速,灵敏度和信息含量高,已应用于变异或突变筛查、病原体快速检测、法医DNA分型等[13-16].关于HRM在菌群分析中的应用,EVERMAN等[17]已有初步探索,WANG等[18]也已将其用于人类唾液菌群的比较,但都要先提取菌群的宏基因组DNA.直接PCR(direct PCR,dPCR)扩增已是成熟的法医DNA分型技术,在菌落筛选、病原体诊断中也有应用[19],可以简化操作、缩短时间,是菌群DNA分析快速化的另一潜在途径.16S rDNA是菌群分析中最常用的标记基因,其V4高变区在进化关系上更接近16S rDNA全长,通用性和分辨率均较高,且在法医学领域已有报道[18,20]. ...
... [18,20]. ...
... 热启动Taq酶、dNTPs、Ex Taq Buffer(Mg2+ free)和MgCl2均购自日本TaKaRa公司,20×EvaGreen荧光染料购自美国Biotium公司,多杀性巴氏杆菌(Pasteurella multocida,Pm)A01菌株的基因组DNA由河南科技大学基因工程疫苗课题组赠送,细菌基因组DNA提取试剂盒购自天根生化科技(北京)有限公司,Tris-EDTA(TE)缓冲液(pH=8.0)购自北京索莱宝科技有限公司.细菌16S rDNA V4区的通用引物[18]由北京博迈德基因技术有限公司合成,序列为:518F(5′-CC AGCAGCCGCGGTAAT-3′)和806R(5′-GGACTACCA GGGTATCTAATCCTGTT-3′). ...
High resolution melting analysis (HRM) based on 16S rRNA as a tool for personal identification with the human oral microbiome
3
2019
... 熔解曲线分析法利用解链温度差异来鉴别DNA,在荧光定量PCR仪上扩增的DNA可直接进行熔解曲线分析[12].使用EvaGreen等饱和荧光染料和高精度的PCR仪,可以提高熔解曲线的分辨率,即高分辨率熔解(high resolution melting,HRM).HRM过程简单、快速,灵敏度和信息含量高,已应用于变异或突变筛查、病原体快速检测、法医DNA分型等[13-16].关于HRM在菌群分析中的应用,EVERMAN等[17]已有初步探索,WANG等[18]也已将其用于人类唾液菌群的比较,但都要先提取菌群的宏基因组DNA.直接PCR(direct PCR,dPCR)扩增已是成熟的法医DNA分型技术,在菌落筛选、病原体诊断中也有应用[19],可以简化操作、缩短时间,是菌群DNA分析快速化的另一潜在途径.16S rDNA是菌群分析中最常用的标记基因,其V4高变区在进化关系上更接近16S rDNA全长,通用性和分辨率均较高,且在法医学领域已有报道[18,20]. ...
... 常规的菌群DNA分析包括检材预处理、DNA提取、扩增和PCR产物分析等,步骤多,耗时长,难以自动化、标准化.其中以DNA提取和产物分析最为繁琐,耗费检材,且可能改变样本菌群结构,增加污染风险.细菌DNA的提取有释放和纯化两个环节,即先用机械、理化或酶解等手段破坏细胞,释放DNA,然后通过抽提或吸附等去除其中杂质.热解法不需要特别的设备、试剂,在各种破坏细胞壁的手段中最为简单.唾液等样本经简单处理后可直接加入PCR体系,利用扩增前预变性时的高温裂解细菌,即可释放出足够的模板[19,22].研究[28]表明,在基于16S rDNA的PCR分析中,各种模板提取方法对结果影响不大,与采样等其他因素相比较几乎可以忽略不计.dPCR技术本身在法医学领域就应用广泛[29-30],如能实现基于dPCR的菌群DNA分析,不仅符合法医学检材特点和检验要求,也更易于普及推广. ...
... 近年已有使用dPCR快速分析细菌DNA的报道[31],但均以特定细菌为目标.与单一序列的扩增不同,菌群PCR不仅需要样本释放足够DNA模板,还应尽量保持各种细菌DNA所占的比例.为了验证dPCR用于菌群分析的可行性,本研究选取5份唾液样本,每份同时使用dPCR-HRM和kPCR-HRM进行菌群分析.两种方法除模板不同外,其他条件保持一致.结果表明,只要两种方法所用模板的样本来源相同,得到的HRM图谱高度相似,GCP超过80%[GCP>95.85%(96.95%±1.56%)].这与多数文献[19,22,28]报道的一致,即提取方法对菌群DNA分析的影响较小.至少对于唾液样本的菌群分型来说,不论直接扩增还是提取后扩增,两者的有效模板构成基本相同,不影响其HRM型的判读.HRM技术用于菌群分析的可行性已被EVERMAN等[17,32]的研究证实,免于提取的dPCR-HRM在此基础上进一步简化,可在90 min内同步完成数十甚至上百个样本的分析,同时也更便于操作的自动化. ...
直接PCR法在口腔牙龈卟啉单胞菌检测中的应用
1
2020
... 熔解曲线分析法利用解链温度差异来鉴别DNA,在荧光定量PCR仪上扩增的DNA可直接进行熔解曲线分析[12].使用EvaGreen等饱和荧光染料和高精度的PCR仪,可以提高熔解曲线的分辨率,即高分辨率熔解(high resolution melting,HRM).HRM过程简单、快速,灵敏度和信息含量高,已应用于变异或突变筛查、病原体快速检测、法医DNA分型等[13-16].关于HRM在菌群分析中的应用,EVERMAN等[17]已有初步探索,WANG等[18]也已将其用于人类唾液菌群的比较,但都要先提取菌群的宏基因组DNA.直接PCR(direct PCR,dPCR)扩增已是成熟的法医DNA分型技术,在菌落筛选、病原体诊断中也有应用[19],可以简化操作、缩短时间,是菌群DNA分析快速化的另一潜在途径.16S rDNA是菌群分析中最常用的标记基因,其V4高变区在进化关系上更接近16S rDNA全长,通用性和分辨率均较高,且在法医学领域已有报道[18,20]. ...
直接PCR法在口腔牙龈卟啉单胞菌检测中的应用
1
2020
... 熔解曲线分析法利用解链温度差异来鉴别DNA,在荧光定量PCR仪上扩增的DNA可直接进行熔解曲线分析[12].使用EvaGreen等饱和荧光染料和高精度的PCR仪,可以提高熔解曲线的分辨率,即高分辨率熔解(high resolution melting,HRM).HRM过程简单、快速,灵敏度和信息含量高,已应用于变异或突变筛查、病原体快速检测、法医DNA分型等[13-16].关于HRM在菌群分析中的应用,EVERMAN等[17]已有初步探索,WANG等[18]也已将其用于人类唾液菌群的比较,但都要先提取菌群的宏基因组DNA.直接PCR(direct PCR,dPCR)扩增已是成熟的法医DNA分型技术,在菌落筛选、病原体诊断中也有应用[19],可以简化操作、缩短时间,是菌群DNA分析快速化的另一潜在途径.16S rDNA是菌群分析中最常用的标记基因,其V4高变区在进化关系上更接近16S rDNA全长,通用性和分辨率均较高,且在法医学领域已有报道[18,20]. ...
16S rRNA基因可变区与全长序列进化关系相似性分析
1
2022
... 按照知情同意原则,采集本校79名志愿者的非刺激性唾液[21]共84份.采集前至少禁食禁水1 h,采集时志愿者身体前倾,收集自然流出的唾液约1 mL于1.5 mL微量离心管中并编号,于采样后30 min内进行后续处理或置于-80 ℃保存备用.其中,1~5号志愿者为本课题组成员(年龄20~60岁;男性3名,女性2名;吸烟者2名,不吸烟者3名),在初次采样3个月后重复采样1次,初次采样样本编号为1~5号,第二次采样样本编号为T1~T5.1号、2号志愿者在第二次采集唾液的同时无菌采集外周静脉血,常规Chelex法提取基因组DNA.其他样本来自本校学生,编号6~79号. ...
16S rRNA基因可变区与全长序列进化关系相似性分析
1
2022
... 按照知情同意原则,采集本校79名志愿者的非刺激性唾液[21]共84份.采集前至少禁食禁水1 h,采集时志愿者身体前倾,收集自然流出的唾液约1 mL于1.5 mL微量离心管中并编号,于采样后30 min内进行后续处理或置于-80 ℃保存备用.其中,1~5号志愿者为本课题组成员(年龄20~60岁;男性3名,女性2名;吸烟者2名,不吸烟者3名),在初次采样3个月后重复采样1次,初次采样样本编号为1~5号,第二次采样样本编号为T1~T5.1号、2号志愿者在第二次采集唾液的同时无菌采集外周静脉血,常规Chelex法提取基因组DNA.其他样本来自本校学生,编号6~79号. ...
高通量测序方法分析实验室培养对人体口腔唾液菌群的影响
3
2018
... 混检样本:分别取编号为T1~T5的唾液样本各50 µL等体积混合,制成可以代表一般个体唾液细菌含量和菌群结构的混检样本[22],采取与唾液样本相同方法处理后,分为2份:1份从3.5倍至3.5×105倍行10倍梯度稀释;另1份用细菌基因组DNA提取试剂盒提取,紫外分光光度法定量后,从250 ng/µL至2.5×10-3 ng/µL行10倍梯度稀释. ...
... 常规的菌群DNA分析包括检材预处理、DNA提取、扩增和PCR产物分析等,步骤多,耗时长,难以自动化、标准化.其中以DNA提取和产物分析最为繁琐,耗费检材,且可能改变样本菌群结构,增加污染风险.细菌DNA的提取有释放和纯化两个环节,即先用机械、理化或酶解等手段破坏细胞,释放DNA,然后通过抽提或吸附等去除其中杂质.热解法不需要特别的设备、试剂,在各种破坏细胞壁的手段中最为简单.唾液等样本经简单处理后可直接加入PCR体系,利用扩增前预变性时的高温裂解细菌,即可释放出足够的模板[19,22].研究[28]表明,在基于16S rDNA的PCR分析中,各种模板提取方法对结果影响不大,与采样等其他因素相比较几乎可以忽略不计.dPCR技术本身在法医学领域就应用广泛[29-30],如能实现基于dPCR的菌群DNA分析,不仅符合法医学检材特点和检验要求,也更易于普及推广. ...
... 近年已有使用dPCR快速分析细菌DNA的报道[31],但均以特定细菌为目标.与单一序列的扩增不同,菌群PCR不仅需要样本释放足够DNA模板,还应尽量保持各种细菌DNA所占的比例.为了验证dPCR用于菌群分析的可行性,本研究选取5份唾液样本,每份同时使用dPCR-HRM和kPCR-HRM进行菌群分析.两种方法除模板不同外,其他条件保持一致.结果表明,只要两种方法所用模板的样本来源相同,得到的HRM图谱高度相似,GCP超过80%[GCP>95.85%(96.95%±1.56%)].这与多数文献[19,22,28]报道的一致,即提取方法对菌群DNA分析的影响较小.至少对于唾液样本的菌群分型来说,不论直接扩增还是提取后扩增,两者的有效模板构成基本相同,不影响其HRM型的判读.HRM技术用于菌群分析的可行性已被EVERMAN等[17,32]的研究证实,免于提取的dPCR-HRM在此基础上进一步简化,可在90 min内同步完成数十甚至上百个样本的分析,同时也更便于操作的自动化. ...
高通量测序方法分析实验室培养对人体口腔唾液菌群的影响
3
2018
... 混检样本:分别取编号为T1~T5的唾液样本各50 µL等体积混合,制成可以代表一般个体唾液细菌含量和菌群结构的混检样本[22],采取与唾液样本相同方法处理后,分为2份:1份从3.5倍至3.5×105倍行10倍梯度稀释;另1份用细菌基因组DNA提取试剂盒提取,紫外分光光度法定量后,从250 ng/µL至2.5×10-3 ng/µL行10倍梯度稀释. ...
... 常规的菌群DNA分析包括检材预处理、DNA提取、扩增和PCR产物分析等,步骤多,耗时长,难以自动化、标准化.其中以DNA提取和产物分析最为繁琐,耗费检材,且可能改变样本菌群结构,增加污染风险.细菌DNA的提取有释放和纯化两个环节,即先用机械、理化或酶解等手段破坏细胞,释放DNA,然后通过抽提或吸附等去除其中杂质.热解法不需要特别的设备、试剂,在各种破坏细胞壁的手段中最为简单.唾液等样本经简单处理后可直接加入PCR体系,利用扩增前预变性时的高温裂解细菌,即可释放出足够的模板[19,22].研究[28]表明,在基于16S rDNA的PCR分析中,各种模板提取方法对结果影响不大,与采样等其他因素相比较几乎可以忽略不计.dPCR技术本身在法医学领域就应用广泛[29-30],如能实现基于dPCR的菌群DNA分析,不仅符合法医学检材特点和检验要求,也更易于普及推广. ...
... 近年已有使用dPCR快速分析细菌DNA的报道[31],但均以特定细菌为目标.与单一序列的扩增不同,菌群PCR不仅需要样本释放足够DNA模板,还应尽量保持各种细菌DNA所占的比例.为了验证dPCR用于菌群分析的可行性,本研究选取5份唾液样本,每份同时使用dPCR-HRM和kPCR-HRM进行菌群分析.两种方法除模板不同外,其他条件保持一致.结果表明,只要两种方法所用模板的样本来源相同,得到的HRM图谱高度相似,GCP超过80%[GCP>95.85%(96.95%±1.56%)].这与多数文献[19,22,28]报道的一致,即提取方法对菌群DNA分析的影响较小.至少对于唾液样本的菌群分型来说,不论直接扩增还是提取后扩增,两者的有效模板构成基本相同,不影响其HRM型的判读.HRM技术用于菌群分析的可行性已被EVERMAN等[17,32]的研究证实,免于提取的dPCR-HRM在此基础上进一步简化,可在90 min内同步完成数十甚至上百个样本的分析,同时也更便于操作的自动化. ...
Considerations and best practices in animal science 16S ribosomal RNA gene sequencing microbiome studies
1
346
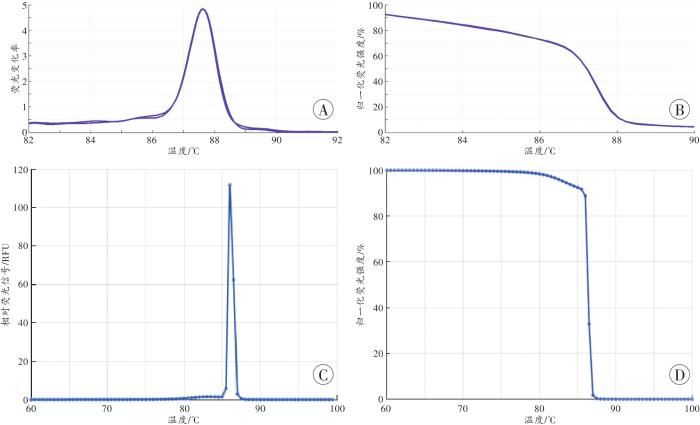
... 以已知序列的阳性对照模板(多杀性巴氏杆菌基因组DNA)优化反应体系、循环参数,建立细菌16S rDNA V4区的PCR-HRM法,所得图谱与uMelt[23](https://dna-utah.org/index.html)预测图谱比较,并用10倍梯度稀释(250 ng/µL至2.5×10-3 ng/µL)的多杀性巴氏杆菌DNA测试灵敏度. ...
Considerations and best practices in animal science 16S ribosomal RNA gene sequencing microbiome studies
1
346
... 以已知序列的阳性对照模板(多杀性巴氏杆菌基因组DNA)优化反应体系、循环参数,建立细菌16S rDNA V4区的PCR-HRM法,所得图谱与uMelt[23](https://dna-utah.org/index.html)预测图谱比较,并用10倍梯度稀释(250 ng/µL至2.5×10-3 ng/µL)的多杀性巴氏杆菌DNA测试灵敏度. ...
uMELT: Prediction of high-resolution melting curves and dynamic melting profiles of PCR products in a rich web application
1
2011
... 参考NUNZIATA等[24-25]的方法,以判型置信度(genotype confidence percentage,GCP)为衡量HRM图谱之间相似性的客观指标.待测样本相对于参考样本的GCP=,其中drt 为待测曲线与参考曲线之间的欧氏距离.应用Rotor-Gene Q 2.3.1软件(德国Qiagen公司),结合人工核查,进行数据分析和判型. ...
Genotype confidence percentage of SSR HRM profiles as a measure of genetic similarity in Rosmarinus officinalis
2
2018
... 参考NUNZIATA等[24-25]的方法,以判型置信度(genotype confidence percentage,GCP)为衡量HRM图谱之间相似性的客观指标.待测样本相对于参考样本的GCP=,其中drt 为待测曲线与参考曲线之间的欧氏距离.应用Rotor-Gene Q 2.3.1软件(德国Qiagen公司),结合人工核查,进行数据分析和判型. ...
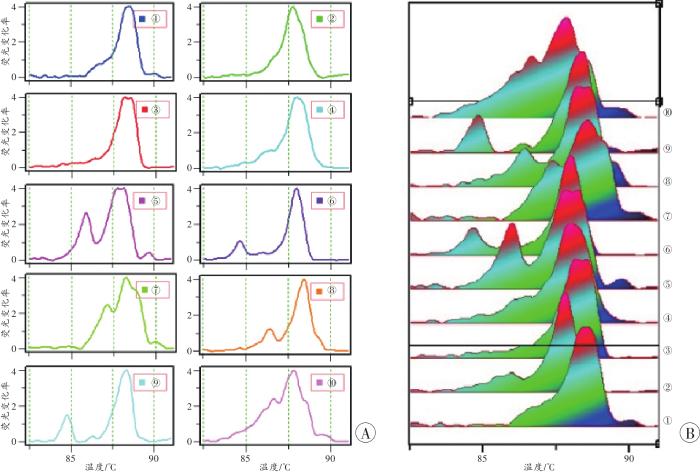
... Rotor-Gene Q 2.3.1软件采集扩增和熔解过程中的荧光强度数据,自动生成扩增曲线和HRM曲线.根据扩增曲线的循环阈值(cycle threshold,Ct)排除扩增失败的反应管;然后用“Melt”模式分析,核查熔解曲线的峰型,确定熔解的温度范围(82.5~91 ℃);再用“HRM”模式分析,以熔解前1 ℃(81.5~82.5 ℃)和后1 ℃(91~92 ℃)两个区间的荧光值为准,对原始HRM曲线进行归一化,生成归一化的HRM曲线.在“HRM”模式下,定义“菌群型”及其参考样本,调整GCP界值,查看待测样本的判型与GCP值.以GCP=80%为界值[26],判断两个HRM图谱是否能归为同一种菌群型.复管取均值后,导出GCP等数据及熔解曲线和HRM图谱.涉及不同批次间的比较,如群体样本调查时,先把上述分批检测、导出的数据用Origin 8软件(美国OriginLab公司)进行整合,各批次均包含22号样本,以控制和修正批次间差异[25],然后导入LabSpec 5软件(日本HORIBA Scientific公司)统一进行平滑、扣基线和校准,生成与系列图谱相应的数据矩阵,再用LabSpec 5软件的“Model”模式进行建模分析,结合数据和人工读图判定新检出的“菌群型”,统计各种型的数目. ...
HRM efficiency and limitations for high-throughput SSR genotyping: A case study using grapevine flavor-linked markers
2
2019
... Rotor-Gene Q 2.3.1软件采集扩增和熔解过程中的荧光强度数据,自动生成扩增曲线和HRM曲线.根据扩增曲线的循环阈值(cycle threshold,Ct)排除扩增失败的反应管;然后用“Melt”模式分析,核查熔解曲线的峰型,确定熔解的温度范围(82.5~91 ℃);再用“HRM”模式分析,以熔解前1 ℃(81.5~82.5 ℃)和后1 ℃(91~92 ℃)两个区间的荧光值为准,对原始HRM曲线进行归一化,生成归一化的HRM曲线.在“HRM”模式下,定义“菌群型”及其参考样本,调整GCP界值,查看待测样本的判型与GCP值.以GCP=80%为界值[26],判断两个HRM图谱是否能归为同一种菌群型.复管取均值后,导出GCP等数据及熔解曲线和HRM图谱.涉及不同批次间的比较,如群体样本调查时,先把上述分批检测、导出的数据用Origin 8软件(美国OriginLab公司)进行整合,各批次均包含22号样本,以控制和修正批次间差异[25],然后导入LabSpec 5软件(日本HORIBA Scientific公司)统一进行平滑、扣基线和校准,生成与系列图谱相应的数据矩阵,再用LabSpec 5软件的“Model”模式进行建模分析,结合数据和人工读图判定新检出的“菌群型”,统计各种型的数目. ...
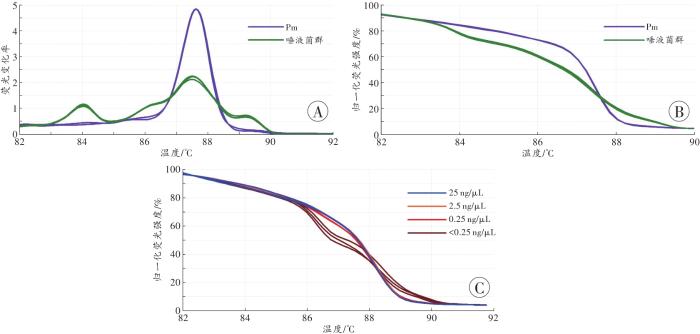
... 菌群结构的多样性或个体差异是菌群分析法医学应用的主要基础.虽然唾液菌群的高度多样性已被包括NGS在内的诸多研究[37-38]证实,但dPCR-HRM所能揭示的多样性尚待调查.本研究结果表明,即使受试者均在生活环境和饮食趋同的高校内,仅16S rDNA V4区就可检出多种类型.且考虑温度、PCR循环次数对HRM分型的影响,参考SAKARIDIS等[26]的研究,本研究选择了较为保守的GCP界值(80%).未来在体系内引入温度内参、控制循环次数,并在增强稳定性的基础上提高GCP界值,有望进一步提高dPCR-HRM技术的分型能力. ...
Genotyping of Listeria monocytogenes isolates from poultry carcasses using high resolution melting (HRM) analysis
1
2014
... 法医学现场检材中细菌的种类和数量包含着其来源个体、遗留时间等信息,为法医学问题的解决提供了新的思路.但目前的菌群分析技术[27]尚难以满足法医学应用要求,需要结合法医学检验目的、技术要求等进行改进和评价.本研究结合dPCR和HRM技术建立了一种可用于人体菌群快速分析的dPCR-HRM技术,并初步评价了其应用于法医学领域的可行性. ...
大鼠尸体细菌群落演替与PMI推断意义
3
2013
... 常规的菌群DNA分析包括检材预处理、DNA提取、扩增和PCR产物分析等,步骤多,耗时长,难以自动化、标准化.其中以DNA提取和产物分析最为繁琐,耗费检材,且可能改变样本菌群结构,增加污染风险.细菌DNA的提取有释放和纯化两个环节,即先用机械、理化或酶解等手段破坏细胞,释放DNA,然后通过抽提或吸附等去除其中杂质.热解法不需要特别的设备、试剂,在各种破坏细胞壁的手段中最为简单.唾液等样本经简单处理后可直接加入PCR体系,利用扩增前预变性时的高温裂解细菌,即可释放出足够的模板[19,22].研究[28]表明,在基于16S rDNA的PCR分析中,各种模板提取方法对结果影响不大,与采样等其他因素相比较几乎可以忽略不计.dPCR技术本身在法医学领域就应用广泛[29-30],如能实现基于dPCR的菌群DNA分析,不仅符合法医学检材特点和检验要求,也更易于普及推广. ...
... 近年已有使用dPCR快速分析细菌DNA的报道[31],但均以特定细菌为目标.与单一序列的扩增不同,菌群PCR不仅需要样本释放足够DNA模板,还应尽量保持各种细菌DNA所占的比例.为了验证dPCR用于菌群分析的可行性,本研究选取5份唾液样本,每份同时使用dPCR-HRM和kPCR-HRM进行菌群分析.两种方法除模板不同外,其他条件保持一致.结果表明,只要两种方法所用模板的样本来源相同,得到的HRM图谱高度相似,GCP超过80%[GCP>95.85%(96.95%±1.56%)].这与多数文献[19,22,28]报道的一致,即提取方法对菌群DNA分析的影响较小.至少对于唾液样本的菌群分型来说,不论直接扩增还是提取后扩增,两者的有效模板构成基本相同,不影响其HRM型的判读.HRM技术用于菌群分析的可行性已被EVERMAN等[17,32]的研究证实,免于提取的dPCR-HRM在此基础上进一步简化,可在90 min内同步完成数十甚至上百个样本的分析,同时也更便于操作的自动化. ...
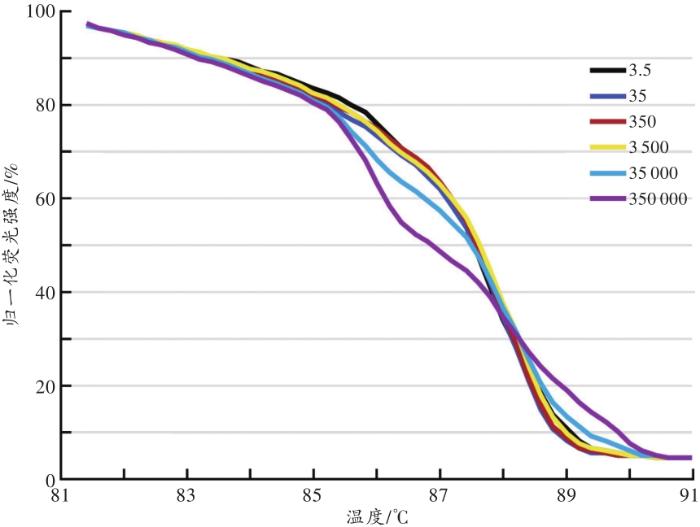
... 为了评价dPCR扩增时菌群模板DNA释放效率及所需最小样本量,本研究同时用dPCR-HRM和kPCR-HRM法调查了一组梯度稀释的唾液样本.该组样本由5人份唾液等体积混合、梯度稀释而成,以降低个体差异影响,更能代表一般个体的情况.调查结果表明,kPCR-HRM法可检测低至2.5×10-3 ng/µL的唾液DNA,dPCR-HRM可检测到2.9×10-3 nL的唾液.根据细菌平均基因组大小(约4×106 bp)[33]和唾液细菌含量(约9×106/µL)[34]估计,两者大约分别相当于500和20~30个细菌.虽然试剂盒提取的唾液DNA中难免混合人和其他微生物的DNA[35],无法准确计算实际的细菌数,但结合两种模板稀释后的唾液量估计,dPCR-HRM法灵敏度不低于kPCR-HRM法,支持菌群DNA可以在本研究的直接扩增条件下有效释放,提取并非必要[28]. ...
大鼠尸体细菌群落演替与PMI推断意义
3
2013
... 常规的菌群DNA分析包括检材预处理、DNA提取、扩增和PCR产物分析等,步骤多,耗时长,难以自动化、标准化.其中以DNA提取和产物分析最为繁琐,耗费检材,且可能改变样本菌群结构,增加污染风险.细菌DNA的提取有释放和纯化两个环节,即先用机械、理化或酶解等手段破坏细胞,释放DNA,然后通过抽提或吸附等去除其中杂质.热解法不需要特别的设备、试剂,在各种破坏细胞壁的手段中最为简单.唾液等样本经简单处理后可直接加入PCR体系,利用扩增前预变性时的高温裂解细菌,即可释放出足够的模板[19,22].研究[28]表明,在基于16S rDNA的PCR分析中,各种模板提取方法对结果影响不大,与采样等其他因素相比较几乎可以忽略不计.dPCR技术本身在法医学领域就应用广泛[29-30],如能实现基于dPCR的菌群DNA分析,不仅符合法医学检材特点和检验要求,也更易于普及推广. ...
... 近年已有使用dPCR快速分析细菌DNA的报道[31],但均以特定细菌为目标.与单一序列的扩增不同,菌群PCR不仅需要样本释放足够DNA模板,还应尽量保持各种细菌DNA所占的比例.为了验证dPCR用于菌群分析的可行性,本研究选取5份唾液样本,每份同时使用dPCR-HRM和kPCR-HRM进行菌群分析.两种方法除模板不同外,其他条件保持一致.结果表明,只要两种方法所用模板的样本来源相同,得到的HRM图谱高度相似,GCP超过80%[GCP>95.85%(96.95%±1.56%)].这与多数文献[19,22,28]报道的一致,即提取方法对菌群DNA分析的影响较小.至少对于唾液样本的菌群分型来说,不论直接扩增还是提取后扩增,两者的有效模板构成基本相同,不影响其HRM型的判读.HRM技术用于菌群分析的可行性已被EVERMAN等[17,32]的研究证实,免于提取的dPCR-HRM在此基础上进一步简化,可在90 min内同步完成数十甚至上百个样本的分析,同时也更便于操作的自动化. ...
... 为了评价dPCR扩增时菌群模板DNA释放效率及所需最小样本量,本研究同时用dPCR-HRM和kPCR-HRM法调查了一组梯度稀释的唾液样本.该组样本由5人份唾液等体积混合、梯度稀释而成,以降低个体差异影响,更能代表一般个体的情况.调查结果表明,kPCR-HRM法可检测低至2.5×10-3 ng/µL的唾液DNA,dPCR-HRM可检测到2.9×10-3 nL的唾液.根据细菌平均基因组大小(约4×106 bp)[33]和唾液细菌含量(约9×106/µL)[34]估计,两者大约分别相当于500和20~30个细菌.虽然试剂盒提取的唾液DNA中难免混合人和其他微生物的DNA[35],无法准确计算实际的细菌数,但结合两种模板稀释后的唾液量估计,dPCR-HRM法灵敏度不低于kPCR-HRM法,支持菌群DNA可以在本研究的直接扩增条件下有效释放,提取并非必要[28]. ...
Evaluation of primer pairs for microbiome profiling from soils to humans within the One Health framework
1
2020
... 常规的菌群DNA分析包括检材预处理、DNA提取、扩增和PCR产物分析等,步骤多,耗时长,难以自动化、标准化.其中以DNA提取和产物分析最为繁琐,耗费检材,且可能改变样本菌群结构,增加污染风险.细菌DNA的提取有释放和纯化两个环节,即先用机械、理化或酶解等手段破坏细胞,释放DNA,然后通过抽提或吸附等去除其中杂质.热解法不需要特别的设备、试剂,在各种破坏细胞壁的手段中最为简单.唾液等样本经简单处理后可直接加入PCR体系,利用扩增前预变性时的高温裂解细菌,即可释放出足够的模板[19,22].研究[28]表明,在基于16S rDNA的PCR分析中,各种模板提取方法对结果影响不大,与采样等其他因素相比较几乎可以忽略不计.dPCR技术本身在法医学领域就应用广泛[29-30],如能实现基于dPCR的菌群DNA分析,不仅符合法医学检材特点和检验要求,也更易于普及推广. ...
直接扩增技术在法医DNA检验中的研究进展
1
2018
... 常规的菌群DNA分析包括检材预处理、DNA提取、扩增和PCR产物分析等,步骤多,耗时长,难以自动化、标准化.其中以DNA提取和产物分析最为繁琐,耗费检材,且可能改变样本菌群结构,增加污染风险.细菌DNA的提取有释放和纯化两个环节,即先用机械、理化或酶解等手段破坏细胞,释放DNA,然后通过抽提或吸附等去除其中杂质.热解法不需要特别的设备、试剂,在各种破坏细胞壁的手段中最为简单.唾液等样本经简单处理后可直接加入PCR体系,利用扩增前预变性时的高温裂解细菌,即可释放出足够的模板[19,22].研究[28]表明,在基于16S rDNA的PCR分析中,各种模板提取方法对结果影响不大,与采样等其他因素相比较几乎可以忽略不计.dPCR技术本身在法医学领域就应用广泛[29-30],如能实现基于dPCR的菌群DNA分析,不仅符合法医学检材特点和检验要求,也更易于普及推广. ...
直接扩增技术在法医DNA检验中的研究进展
1
2018
... 常规的菌群DNA分析包括检材预处理、DNA提取、扩增和PCR产物分析等,步骤多,耗时长,难以自动化、标准化.其中以DNA提取和产物分析最为繁琐,耗费检材,且可能改变样本菌群结构,增加污染风险.细菌DNA的提取有释放和纯化两个环节,即先用机械、理化或酶解等手段破坏细胞,释放DNA,然后通过抽提或吸附等去除其中杂质.热解法不需要特别的设备、试剂,在各种破坏细胞壁的手段中最为简单.唾液等样本经简单处理后可直接加入PCR体系,利用扩增前预变性时的高温裂解细菌,即可释放出足够的模板[19,22].研究[28]表明,在基于16S rDNA的PCR分析中,各种模板提取方法对结果影响不大,与采样等其他因素相比较几乎可以忽略不计.dPCR技术本身在法医学领域就应用广泛[29-30],如能实现基于dPCR的菌群DNA分析,不仅符合法医学检材特点和检验要求,也更易于普及推广. ...
擦拭直扩法和粘取磁珠法检测衣物脱落细胞DNA的比较
1
2017
... 近年已有使用dPCR快速分析细菌DNA的报道[31],但均以特定细菌为目标.与单一序列的扩增不同,菌群PCR不仅需要样本释放足够DNA模板,还应尽量保持各种细菌DNA所占的比例.为了验证dPCR用于菌群分析的可行性,本研究选取5份唾液样本,每份同时使用dPCR-HRM和kPCR-HRM进行菌群分析.两种方法除模板不同外,其他条件保持一致.结果表明,只要两种方法所用模板的样本来源相同,得到的HRM图谱高度相似,GCP超过80%[GCP>95.85%(96.95%±1.56%)].这与多数文献[19,22,28]报道的一致,即提取方法对菌群DNA分析的影响较小.至少对于唾液样本的菌群分型来说,不论直接扩增还是提取后扩增,两者的有效模板构成基本相同,不影响其HRM型的判读.HRM技术用于菌群分析的可行性已被EVERMAN等[17,32]的研究证实,免于提取的dPCR-HRM在此基础上进一步简化,可在90 min内同步完成数十甚至上百个样本的分析,同时也更便于操作的自动化. ...
擦拭直扩法和粘取磁珠法检测衣物脱落细胞DNA的比较
1
2017
... 近年已有使用dPCR快速分析细菌DNA的报道[31],但均以特定细菌为目标.与单一序列的扩增不同,菌群PCR不仅需要样本释放足够DNA模板,还应尽量保持各种细菌DNA所占的比例.为了验证dPCR用于菌群分析的可行性,本研究选取5份唾液样本,每份同时使用dPCR-HRM和kPCR-HRM进行菌群分析.两种方法除模板不同外,其他条件保持一致.结果表明,只要两种方法所用模板的样本来源相同,得到的HRM图谱高度相似,GCP超过80%[GCP>95.85%(96.95%±1.56%)].这与多数文献[19,22,28]报道的一致,即提取方法对菌群DNA分析的影响较小.至少对于唾液样本的菌群分型来说,不论直接扩增还是提取后扩增,两者的有效模板构成基本相同,不影响其HRM型的判读.HRM技术用于菌群分析的可行性已被EVERMAN等[17,32]的研究证实,免于提取的dPCR-HRM在此基础上进一步简化,可在90 min内同步完成数十甚至上百个样本的分析,同时也更便于操作的自动化. ...
Direct PCR amplification from saliva sample using non-direct multiplex STR kits for forensic DNA typing
1
2021
... 近年已有使用dPCR快速分析细菌DNA的报道[31],但均以特定细菌为目标.与单一序列的扩增不同,菌群PCR不仅需要样本释放足够DNA模板,还应尽量保持各种细菌DNA所占的比例.为了验证dPCR用于菌群分析的可行性,本研究选取5份唾液样本,每份同时使用dPCR-HRM和kPCR-HRM进行菌群分析.两种方法除模板不同外,其他条件保持一致.结果表明,只要两种方法所用模板的样本来源相同,得到的HRM图谱高度相似,GCP超过80%[GCP>95.85%(96.95%±1.56%)].这与多数文献[19,22,28]报道的一致,即提取方法对菌群DNA分析的影响较小.至少对于唾液样本的菌群分型来说,不论直接扩增还是提取后扩增,两者的有效模板构成基本相同,不影响其HRM型的判读.HRM技术用于菌群分析的可行性已被EVERMAN等[17,32]的研究证实,免于提取的dPCR-HRM在此基础上进一步简化,可在90 min内同步完成数十甚至上百个样本的分析,同时也更便于操作的自动化. ...
High-resolution melt analysis for rapid comparison of bacterial community compositions
1
03923
... 为了评价dPCR扩增时菌群模板DNA释放效率及所需最小样本量,本研究同时用dPCR-HRM和kPCR-HRM法调查了一组梯度稀释的唾液样本.该组样本由5人份唾液等体积混合、梯度稀释而成,以降低个体差异影响,更能代表一般个体的情况.调查结果表明,kPCR-HRM法可检测低至2.5×10-3 ng/µL的唾液DNA,dPCR-HRM可检测到2.9×10-3 nL的唾液.根据细菌平均基因组大小(约4×106 bp)[33]和唾液细菌含量(约9×106/µL)[34]估计,两者大约分别相当于500和20~30个细菌.虽然试剂盒提取的唾液DNA中难免混合人和其他微生物的DNA[35],无法准确计算实际的细菌数,但结合两种模板稀释后的唾液量估计,dPCR-HRM法灵敏度不低于kPCR-HRM法,支持菌群DNA可以在本研究的直接扩增条件下有效释放,提取并非必要[28]. ...
High-resolution melt analysis for rapid comparison of bacterial community compositions
1
03923
... 为了评价dPCR扩增时菌群模板DNA释放效率及所需最小样本量,本研究同时用dPCR-HRM和kPCR-HRM法调查了一组梯度稀释的唾液样本.该组样本由5人份唾液等体积混合、梯度稀释而成,以降低个体差异影响,更能代表一般个体的情况.调查结果表明,kPCR-HRM法可检测低至2.5×10-3 ng/µL的唾液DNA,dPCR-HRM可检测到2.9×10-3 nL的唾液.根据细菌平均基因组大小(约4×106 bp)[33]和唾液细菌含量(约9×106/µL)[34]估计,两者大约分别相当于500和20~30个细菌.虽然试剂盒提取的唾液DNA中难免混合人和其他微生物的DNA[35],无法准确计算实际的细菌数,但结合两种模板稀释后的唾液量估计,dPCR-HRM法灵敏度不低于kPCR-HRM法,支持菌群DNA可以在本研究的直接扩增条件下有效释放,提取并非必要[28]. ...
adb1758
1
2022
... 为了评价dPCR扩增时菌群模板DNA释放效率及所需最小样本量,本研究同时用dPCR-HRM和kPCR-HRM法调查了一组梯度稀释的唾液样本.该组样本由5人份唾液等体积混合、梯度稀释而成,以降低个体差异影响,更能代表一般个体的情况.调查结果表明,kPCR-HRM法可检测低至2.5×10-3 ng/µL的唾液DNA,dPCR-HRM可检测到2.9×10-3 nL的唾液.根据细菌平均基因组大小(约4×106 bp)[33]和唾液细菌含量(约9×106/µL)[34]估计,两者大约分别相当于500和20~30个细菌.虽然试剂盒提取的唾液DNA中难免混合人和其他微生物的DNA[35],无法准确计算实际的细菌数,但结合两种模板稀释后的唾液量估计,dPCR-HRM法灵敏度不低于kPCR-HRM法,支持菌群DNA可以在本研究的直接扩增条件下有效释放,提取并非必要[28]. ...
adb1758
1
2022
... 为了评价dPCR扩增时菌群模板DNA释放效率及所需最小样本量,本研究同时用dPCR-HRM和kPCR-HRM法调查了一组梯度稀释的唾液样本.该组样本由5人份唾液等体积混合、梯度稀释而成,以降低个体差异影响,更能代表一般个体的情况.调查结果表明,kPCR-HRM法可检测低至2.5×10-3 ng/µL的唾液DNA,dPCR-HRM可检测到2.9×10-3 nL的唾液.根据细菌平均基因组大小(约4×106 bp)[33]和唾液细菌含量(约9×106/µL)[34]估计,两者大约分别相当于500和20~30个细菌.虽然试剂盒提取的唾液DNA中难免混合人和其他微生物的DNA[35],无法准确计算实际的细菌数,但结合两种模板稀释后的唾液量估计,dPCR-HRM法灵敏度不低于kPCR-HRM法,支持菌群DNA可以在本研究的直接扩增条件下有效释放,提取并非必要[28]. ...
2
2001
... 为了评价dPCR扩增时菌群模板DNA释放效率及所需最小样本量,本研究同时用dPCR-HRM和kPCR-HRM法调查了一组梯度稀释的唾液样本.该组样本由5人份唾液等体积混合、梯度稀释而成,以降低个体差异影响,更能代表一般个体的情况.调查结果表明,kPCR-HRM法可检测低至2.5×10-3 ng/µL的唾液DNA,dPCR-HRM可检测到2.9×10-3 nL的唾液.根据细菌平均基因组大小(约4×106 bp)[33]和唾液细菌含量(约9×106/µL)[34]估计,两者大约分别相当于500和20~30个细菌.虽然试剂盒提取的唾液DNA中难免混合人和其他微生物的DNA[35],无法准确计算实际的细菌数,但结合两种模板稀释后的唾液量估计,dPCR-HRM法灵敏度不低于kPCR-HRM法,支持菌群DNA可以在本研究的直接扩增条件下有效释放,提取并非必要[28]. ...
... 本研究建立了一种不需要提取和电泳的dPCR-HRM菌群分析技术,简单快速且有较高的灵敏度、稳定性和鉴别能力,并初步验证了其在法医学领域的应用.但是,必须意识到菌群分析是以群体特征为目标的微生态学研究,菌群PCR与传统的单基因座PCR、复合扩增均有诸多区别.本研究仅以16S rDNA V4区和唾液(斑)为例,且只进行了小样本评估,包含温度内参的多片段dPCR-HRM分析、dPCR-HRM用于其他类型体液(斑)及应对复杂检材的能力还有待进一步研究.现场斑痕的溯源是法医学的核心问题,虽然人体菌群有高度的个体特异性,但目前还难以通过菌群分析进行个体识别.未来应扩大样本范围和样本量,调查不同采样时机和方法,全面评价各种影响因素,结合菌群溯源技术发展[35,41],积极探索实现法医学斑痕菌群溯源的途径. ...
2
2001
... 为了评价dPCR扩增时菌群模板DNA释放效率及所需最小样本量,本研究同时用dPCR-HRM和kPCR-HRM法调查了一组梯度稀释的唾液样本.该组样本由5人份唾液等体积混合、梯度稀释而成,以降低个体差异影响,更能代表一般个体的情况.调查结果表明,kPCR-HRM法可检测低至2.5×10-3 ng/µL的唾液DNA,dPCR-HRM可检测到2.9×10-3 nL的唾液.根据细菌平均基因组大小(约4×106 bp)[33]和唾液细菌含量(约9×106/µL)[34]估计,两者大约分别相当于500和20~30个细菌.虽然试剂盒提取的唾液DNA中难免混合人和其他微生物的DNA[35],无法准确计算实际的细菌数,但结合两种模板稀释后的唾液量估计,dPCR-HRM法灵敏度不低于kPCR-HRM法,支持菌群DNA可以在本研究的直接扩增条件下有效释放,提取并非必要[28]. ...
... 本研究建立了一种不需要提取和电泳的dPCR-HRM菌群分析技术,简单快速且有较高的灵敏度、稳定性和鉴别能力,并初步验证了其在法医学领域的应用.但是,必须意识到菌群分析是以群体特征为目标的微生态学研究,菌群PCR与传统的单基因座PCR、复合扩增均有诸多区别.本研究仅以16S rDNA V4区和唾液(斑)为例,且只进行了小样本评估,包含温度内参的多片段dPCR-HRM分析、dPCR-HRM用于其他类型体液(斑)及应对复杂检材的能力还有待进一步研究.现场斑痕的溯源是法医学的核心问题,虽然人体菌群有高度的个体特异性,但目前还难以通过菌群分析进行个体识别.未来应扩大样本范围和样本量,调查不同采样时机和方法,全面评价各种影响因素,结合菌群溯源技术发展[35,41],积极探索实现法医学斑痕菌群溯源的途径. ...
Integrating the human microbiome in the forensic toolkit: Current bottlenecks and future solutions
1
2022
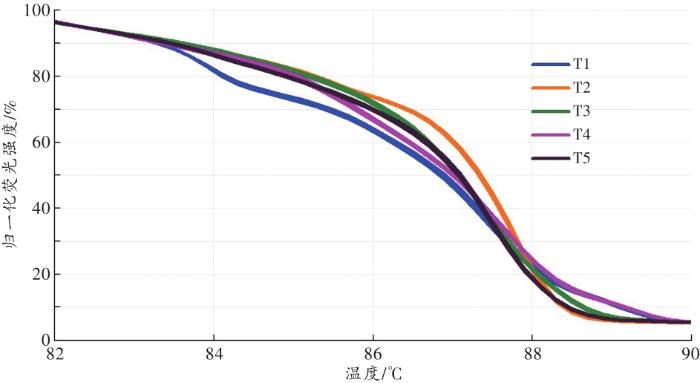
... 理想的菌群快速检测还应有较好的鉴别能力、稳定性和检材适应性[36].为了进一步验证dPCR-HRM鉴别菌群的能力,评价其稳定性和检材适应性,本研究调查了一组唾液样本和唾液斑样本.结果表明,来自不同个体的唾液有不同的HRM图谱.图谱的个体间差异虽然大小不一[GCP<87.07%(52.11%±22.79%)],但远大于同一样本在不同时间检测时的差异[GCP>92.39%(97.78%±2.30%)].且同一样本的dPCR-HRM图谱与kPCR-HRM图谱基本相同,方法间的差异也远小于样本间的差异.虽然样本量有限,但支持dPCR-HRM有较好的鉴别能力和稳定性. ...
微生物法医学的研究现状与进展
1
2012
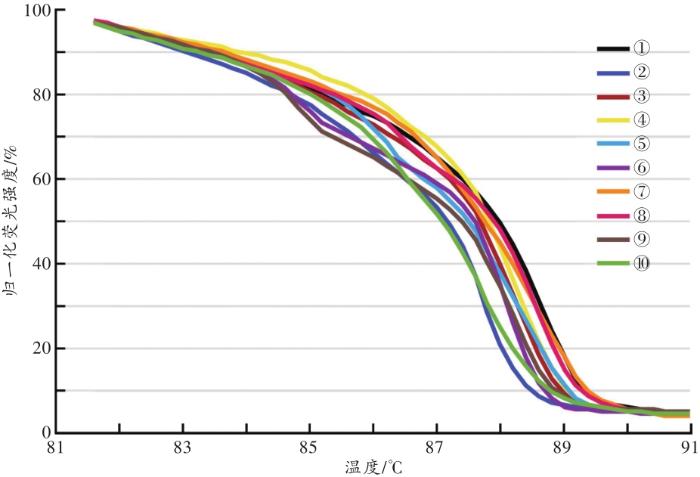
... 菌群结构的多样性或个体差异是菌群分析法医学应用的主要基础.虽然唾液菌群的高度多样性已被包括NGS在内的诸多研究[37-38]证实,但dPCR-HRM所能揭示的多样性尚待调查.本研究结果表明,即使受试者均在生活环境和饮食趋同的高校内,仅16S rDNA V4区就可检出多种类型.且考虑温度、PCR循环次数对HRM分型的影响,参考SAKARIDIS等[26]的研究,本研究选择了较为保守的GCP界值(80%).未来在体系内引入温度内参、控制循环次数,并在增强稳定性的基础上提高GCP界值,有望进一步提高dPCR-HRM技术的分型能力. ...
微生物法医学的研究现状与进展
1
2012
... 菌群结构的多样性或个体差异是菌群分析法医学应用的主要基础.虽然唾液菌群的高度多样性已被包括NGS在内的诸多研究[37-38]证实,但dPCR-HRM所能揭示的多样性尚待调查.本研究结果表明,即使受试者均在生活环境和饮食趋同的高校内,仅16S rDNA V4区就可检出多种类型.且考虑温度、PCR循环次数对HRM分型的影响,参考SAKARIDIS等[26]的研究,本研究选择了较为保守的GCP界值(80%).未来在体系内引入温度内参、控制循环次数,并在增强稳定性的基础上提高GCP界值,有望进一步提高dPCR-HRM技术的分型能力. ...
Oral cavity contains distinct niches with dynamic microbial communities
1
2015
... 菌群结构的多样性或个体差异是菌群分析法医学应用的主要基础.虽然唾液菌群的高度多样性已被包括NGS在内的诸多研究[37-38]证实,但dPCR-HRM所能揭示的多样性尚待调查.本研究结果表明,即使受试者均在生活环境和饮食趋同的高校内,仅16S rDNA V4区就可检出多种类型.且考虑温度、PCR循环次数对HRM分型的影响,参考SAKARIDIS等[26]的研究,本研究选择了较为保守的GCP界值(80%).未来在体系内引入温度内参、控制循环次数,并在增强稳定性的基础上提高GCP界值,有望进一步提高dPCR-HRM技术的分型能力. ...
Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples
1
2012
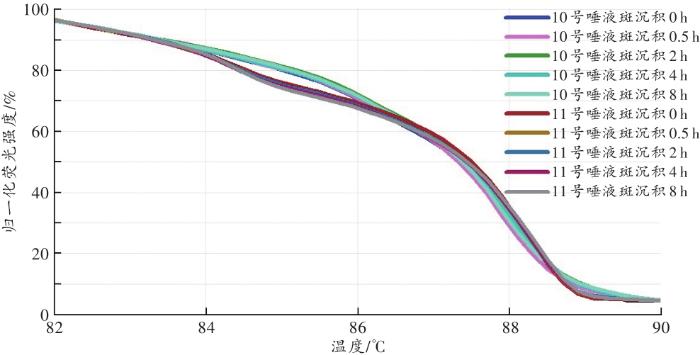
... 检材的复杂性是法医学检验的显著特征,斑痕是最常见的现场检材.本研究对dPCR-HRM用于唾液斑的可行性进行了初步评估,发现模拟唾液斑用常规的纱线转移法即可进行菌群分析.结合上述灵敏度实验,支持dPCR-HRM菌群检验并不需要额外的检材处理和消耗,多数情况下常规检材浸洗液即可满足要求.对不同沉积时间唾液斑的调查结果表明,8 h之内唾液斑与其来源样本的图谱基本相同[GCP>90.83%(96.67%±2.98%)],说明本方法所揭示的斑痕菌群结构是相对稳定的,至少与形成斑痕时的唾液有可比性,这与关于采样时机、采集方法均会影响NGS菌群分析的报道[39]并不一致.结合文献[40]分析认为,与NGS的高成本、高分辨率不同,本研究所述方法类似于经典的DGGE菌群分析,分辨率低,对属以下水平变化、低丰度菌属变化和幅度小的变化不敏感.同时,由于唾液含菌量大,高丰度菌属间的比例不易因污染、采样而大幅改变.因此,唾液菌群的HRM图谱相对稳定,但该方法的分辨率或图谱的个体特异性远小于NGS分析,其意义主要在于样本初筛,在个体识别方面,可通过HRM图谱比较快速地排除一部分样本,为重点样本的精准分析创造条件,弥补NGS成本高、耗时长的局限性.如用于含菌量低、优势菌属不明显的样本,应控制环境、试剂和耗材等可能的细菌污染源,统一采样方式和提取方法,以保证样本间的可比性. ...
Towards standards for human fecal sample processing in metagenomic studies
1
2017
... 检材的复杂性是法医学检验的显著特征,斑痕是最常见的现场检材.本研究对dPCR-HRM用于唾液斑的可行性进行了初步评估,发现模拟唾液斑用常规的纱线转移法即可进行菌群分析.结合上述灵敏度实验,支持dPCR-HRM菌群检验并不需要额外的检材处理和消耗,多数情况下常规检材浸洗液即可满足要求.对不同沉积时间唾液斑的调查结果表明,8 h之内唾液斑与其来源样本的图谱基本相同[GCP>90.83%(96.67%±2.98%)],说明本方法所揭示的斑痕菌群结构是相对稳定的,至少与形成斑痕时的唾液有可比性,这与关于采样时机、采集方法均会影响NGS菌群分析的报道[39]并不一致.结合文献[40]分析认为,与NGS的高成本、高分辨率不同,本研究所述方法类似于经典的DGGE菌群分析,分辨率低,对属以下水平变化、低丰度菌属变化和幅度小的变化不敏感.同时,由于唾液含菌量大,高丰度菌属间的比例不易因污染、采样而大幅改变.因此,唾液菌群的HRM图谱相对稳定,但该方法的分辨率或图谱的个体特异性远小于NGS分析,其意义主要在于样本初筛,在个体识别方面,可通过HRM图谱比较快速地排除一部分样本,为重点样本的精准分析创造条件,弥补NGS成本高、耗时长的局限性.如用于含菌量低、优势菌属不明显的样本,应控制环境、试剂和耗材等可能的细菌污染源,统一采样方式和提取方法,以保证样本间的可比性. ...
A practical guide to amplicon and metagenomic analysis of microbiome data
1
2021
... 本研究建立了一种不需要提取和电泳的dPCR-HRM菌群分析技术,简单快速且有较高的灵敏度、稳定性和鉴别能力,并初步验证了其在法医学领域的应用.但是,必须意识到菌群分析是以群体特征为目标的微生态学研究,菌群PCR与传统的单基因座PCR、复合扩增均有诸多区别.本研究仅以16S rDNA V4区和唾液(斑)为例,且只进行了小样本评估,包含温度内参的多片段dPCR-HRM分析、dPCR-HRM用于其他类型体液(斑)及应对复杂检材的能力还有待进一步研究.现场斑痕的溯源是法医学的核心问题,虽然人体菌群有高度的个体特异性,但目前还难以通过菌群分析进行个体识别.未来应扩大样本范围和样本量,调查不同采样时机和方法,全面评价各种影响因素,结合菌群溯源技术发展[35,41],积极探索实现法医学斑痕菌群溯源的途径. ...
Limitation of high-resolution melting curve analysis for genotyping simple sequence repeats in sheep
0
2014



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}